现在随着三代测序价格跟重测序价格的持续走低,越来越多的全基因组等级的群体遗传文章持续发布。而当拿到一批raw data之后,通常是需要经过一系列是上游分析处理使之成为可以操作的下游raw data,这个过程就是我们经常说的snp calling的过程。对于snp是如何得到的,这里我推荐基因所明哥的一篇博文。
https://ming-lian.github.io/2019/02/08/call-snp/
该篇博文将gatk的流程说得很透彻,各位如果需要可以仔细研读一番。
对于模式生物,我们可以使用VQSR的方法进行过滤。但对于非模式生物,没有现成的snp库,通常我们都是使用hard-filter的方式进行过滤,对于参数的选择,gatk的官方给出了推荐参数,但往往推荐参数并不适合我们手头上的数据类型。如果snp过滤不当,下游的分析会异常难以进行(特指这个倒霉催返回来重新过滤一遍的我)。
在明哥的博文里面,我们可以看到这样的几张图
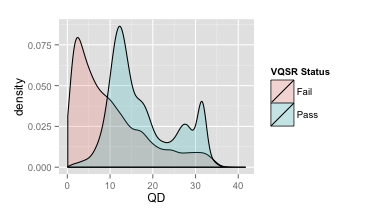
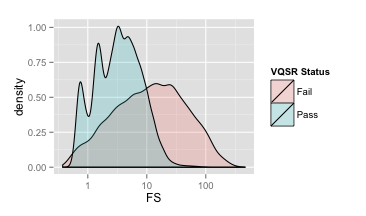
这几张图便是我们进行过滤参数选择的主要依据,掌握好这几张图的做法,对于我们过滤的时候至关重要。
这里介绍的方法是我这两天的劳动成果,在这里再次感谢无私的鲍大哥带我跨过坑。
第一步,我们需要从raw vcf文件中得到我们需要的信息,这里以提取QD这个值为例。
###使用bcftools的query模块进行提取(提取FS就将QD改为FS即可)
bcftools query -f "%QD\n" raw_snp.vcf > my_snp.QD
这样我们就得到了vcf文件中的QD值,接着我们需要把提取得到的QD值里面的.去掉,不然在作图的时候会提示报错,这里我使用python进行过滤
###vim clean_point.py
import sys
x = sys.argv[1]
file = open(x, "r")
lines = file.readlines()
for line in lines:
if str(line) != ".\n":
print(line.rstrip())
###python clean_point.py my_snp.QD > clean_snp.QD
完成过滤之后,我们就可以导进r或者python作图,这里两种方法都介绍一下
首先是python(本操作在jupyter里面进行)
###引入模块
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
###导入数据
df = pd.read_csv("clean_snp.QD",names="Q")
sns.kdeplot(df["Q"], shade=True)
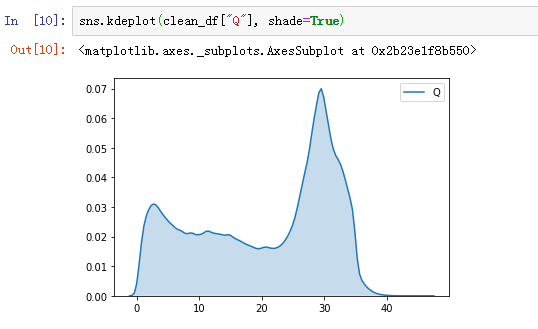
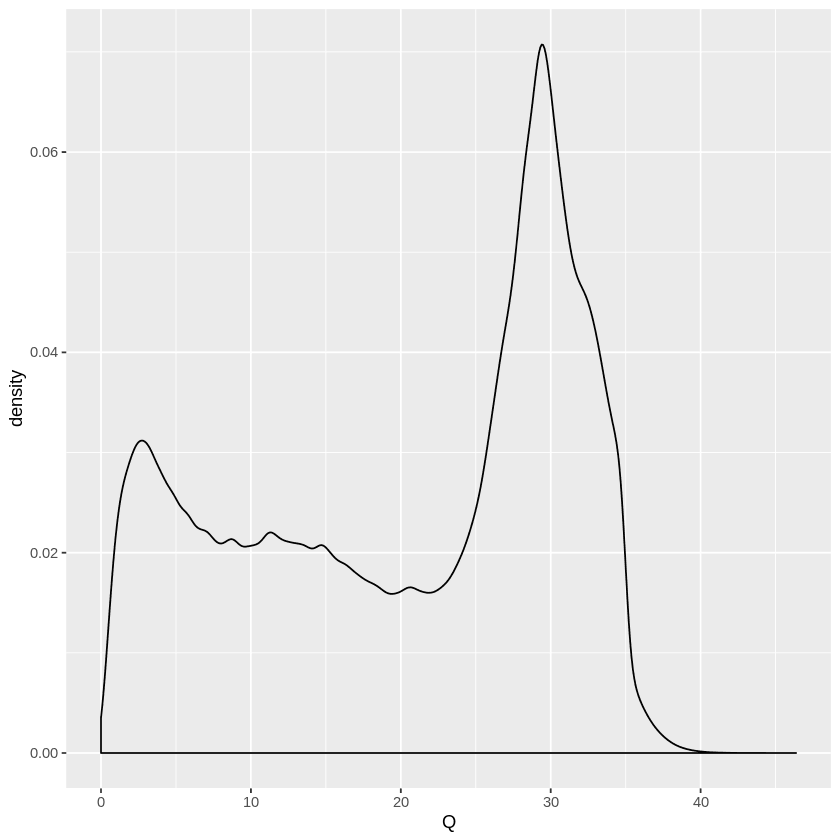
接着就是R
library(ggplot2)
snp_QD <- read.table("clean_snp.QD",header = FALSE)
names(snp_QD) <- ("Q")
ggplot(aes(x=Q###此处就是你要输入的那一列的列名,不要加双引号,我早上就是因为加双引号没出图,后面是潜哥帮我指出错误),data = snp_QD) + geom_density()
其他的也是照着画就行,最后还是再次感谢这两天帮过我的各位~